Structural Variant Calling From NGS Data

Single Nucleotide Variant (SNVs) have been considered as the main source of genetic variation, therefore precisely identifying these SNVs is a critical part of the Next Generation Sequencing (NGS) workflow. However, in this report from 2004, the authors identified another form of variants called the Structural Variants (SVs), which are genetic alterations of 50 or more base pairs, and result in duplications, deletions, insertions, inversions, and translocations in the genome. The changes in the DNA organization resulting from these SVs have been shown to be responsible for both phenotypic variation and a variety of pathological conditions. While the average variation, as measured for SNV is about 0.1% between two individuals; when the SVs are included, the variation rises to approximately 1.5%. Therefore, understanding how to identify and characterize the SVs using NGS data is critical.
Detection techniques for SVs are based on array comparative genomic hybridization (aCGH); very useful in detecting Copy Number Variations (CNVs) and single-nucleotide polymorphism arrays. NGS has enabled methods for the precise definition of SVs breakpoints of different sizes and types. However, due to the volume of the data and short sequence reads obtained from high-throughput sequencing the classification of SVs presents significant challenges.
Deletions, duplications, inversions, and translocations are the different types of SVs that occur due to chromosomal rearrangements. Copy number variations (CNVs) are a particular subtype of SVs mainly represented by deletions and duplications. By using the coverage method, deletion and duplication CNVs can be easily detected from the NGS sequence data, however, inversions are copy number-neutral, and hence coverage method cannot be used in its identification. Translocation is the exchange of genetic material between two non-homologous chromosomes. There are two main types of translocations: reciprocal and Robertsonian. In reciprocal translocation, two different chromosomes exchange segments with each other, whereas, in a Robertsonian translocation, an entire chromosome gets attached to another at the centromere.
NGS produces millions of reads in a single sequencing run by parallelizing the sequencing process. Consequently, it has provided a significant contribution to the detection of Single Nucleotide Polymorphism (SNP). However, since the nature of NGS sequencing is to produce short-read sequences (50 – 400 base pairs), the longer variants (SVs) remain poorly characterized. Due to the relevance of SVs in interpreting phenotypic variability and diseases by affecting more bases than SNPs, it is considered an important genetic variation. As a result, efforts have been made to develop algorithms to address the issue of structural variants calling from NGS data.
With recent technological and methodological developments, long-read sequencing technology, such as Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) are capable of producing reads of several thousand base pairs, even reaching up to 2 Mbp for Oxford Nanopore. Although PacBio and Oxford Nanopore methods can generate reads of thousands of base pairs, they pose two major disadvantages. First, the cost of sequencing is higher to obtain the same coverage compared to short-read sequencing. Second, the sequencing error rate is high – about 8 – 20% – which has to be considered for both alignment and SV calling steps. In spite of the drawbacks, long reads are advantageous for SV calling because they can span repetitive regions, improve the mapping, and capture large SVs better than the short-reads.
As we can fathom, both short-read and long-read sequence data come with their advantages and disadvantages for SV detection. Hence, bioinformaticians and researchers developed algorithms appropriate to the produced sequence reads for the SVs to be efficiently determined and characterized. Here, I am mentioning a few of the important algorithms/tools and their methodologies that are widely used for SV and CNV detection.
Algorithms for SV detection depend on various properties of the underlying sequence data and differ in accuracy and sensitivity. These algorithms follow one or more methods that can be divided into categories:
(1) read depth (RD)
(2) pairing end (PE)
(3) split reads (SR)
(4) de novo assembly (AS)
The most effective approach for the identification of SV depends on the size and the type of the variant as well as the sequencing data characteristics. It is recommended to use multiple variant callers in the SV detection phase.
Variant callers take in a BAM/FastQ file and return a list of variants. Some of the widely used tools for SV, SNP, and Indels include:
- Genome Analysis Toolkit HaplotypeCaller (GATK-HC)
- Samtools mpileup
- Freebayes
Annotation of the identified variants can be done using tools such as:
There are other comprehensive pipelines to systematically handle a large number of variants, which include.
With this information, let’s turn our attention to some of the significant and widely implemented algorithms currently being used for SV calling.
1. De novo assembly-based algorithm
I have elaborately explained the essential concepts in genome assembly in one of my previous blogs. Here, we will concentrate on understanding how de novo assembled sequences can be used to detect SVs/CNVs. In this method, the assembled sequences are aligned to the reference genome or other assemblies, and the aberrations between the two are determined. With the comparison between the genomic positions in the reference and the assembled sequence, different types of variations and discontinuity patterns are identified. It is to be noted that the alignment of such large size sequences is not a computationally trivial task. Conventional matrix-based alignment approaches could lead to out of memory errors – this limiting behavior of a function when the argument tends towards a particular value or infinity is called Big O Notation. To circumvent the issue, a few aligners have been developed, such as:
Most of these aligners like Mummer use a suffix tree data structure approach, and is one of the fastest and most efficient systems available for this task, enabling it to be applied to very long sequences. Some efficient applications using these aligners are:
These applications are used on previously assembled contigs or scaffolds i.e the sequence data has already been assembled, and only the alignment needs to be performed to detect the SVs.
For the case where the detections of SVs have to be executed on unassembled sequence data, de Bruijn graph (refer to blog) and string graph-based approaches are widely used as they leverage the sequence read information directly. Cortex is one of these methods that use short-read sequencing data to assemble several genomes using the de Bruijn graph approach and infer SVs and complex combinations of indels, SNVs, and rearrangements.
SGVar is another method that uses the string graph-based approach to assemble short-read sequence data. SGVar executes stringent assembly processing by taking read length and read quality into account. Additionally, it requires a perfect match of the sequence reads to be merged, which improves the assembly quality. Due to these reasons, SGVar has been shown to outperform Cortex for the identification of insertions and deletions.
Compared with other methods, the key strength of the de novo assembly-based approach lies in detecting larger insertions (up to 3+ kbp). However, the absence of haplotype representation is one significant problem. Consequently, heterozygous SVs are often missed because a de novo assembly represents one haplotype. Nevertheless, there are methods to counter this problem, such as:
Having said that, the de novo assembly approach should be used for a small number of samples and for studying organisms that do not have a reference genome available.
2. Paired-end alignment approach
Paired-end sequence reads typically map in the opposite direction (refer to blog) and within a certain distance of each other, for example, 500 base pairs. This very characteristic of paired-end reads allows the detection of different types of structural variants. In this approach, variations are detected by comparing the expected insert size between the paired-end reads and the observed insert size in the alignment. In the presence of SVs, these reads are abnormally oriented and spaced; reads map at inconsistent distances in the alignment denoting insertions, deletions, or inversions. Discordantly mapped paired reads can be:
(a) further apart than expected
(b) closer together than expected
(c) in incorrect order
(d) in inverse orientation
(e) on different chromosomes.
Over the years, quite a few short-read-based mappers have been introduced. The methods for detecting SVs from short reads differ in the type of information they exploit. BreakDancer is one of the important short-read mapper methods, which classifies the reads into normal or SV depending on the orientation and mapping distance between the read and its mate. The Paired-end approach could detect breakpoints (Figure 1). The resolution of the breakpoints depends on the insert size and the read coverage. In such a scenario, split-reads can be used to detect SVs with a single base-pair resolution. These split-reads contain the breakpoint of the structural variant. Alignments of the split-reads to the reference genome are split into two parts as shown in Figure 1. Parts of a split-read are independently aligned to the reference genome, hence the reads should be long enough to be aligned uniquely. DELLY is a robust method involving split-reads approach that can be used to integrate the analysis of split reads and to search for abnormal distances and orientations among pairs of reads. This increases the accuracy of breakpoint prediction and enables the detection of smaller deletions (20+ bp), however, the larger events remain hard to be distinguished from the mapping artifacts. To overcome this issue, coverage information can be taken into consideration to further improve SV detection.
LUMPY and Manta use all three aspects of the read-depth/coverage, paired-end read discordance, and split-reads to perform analysis. Furthermore, to detect more complex events such as a tandem duplication where the second copy is inverted, methods such as TARDIS can be used.
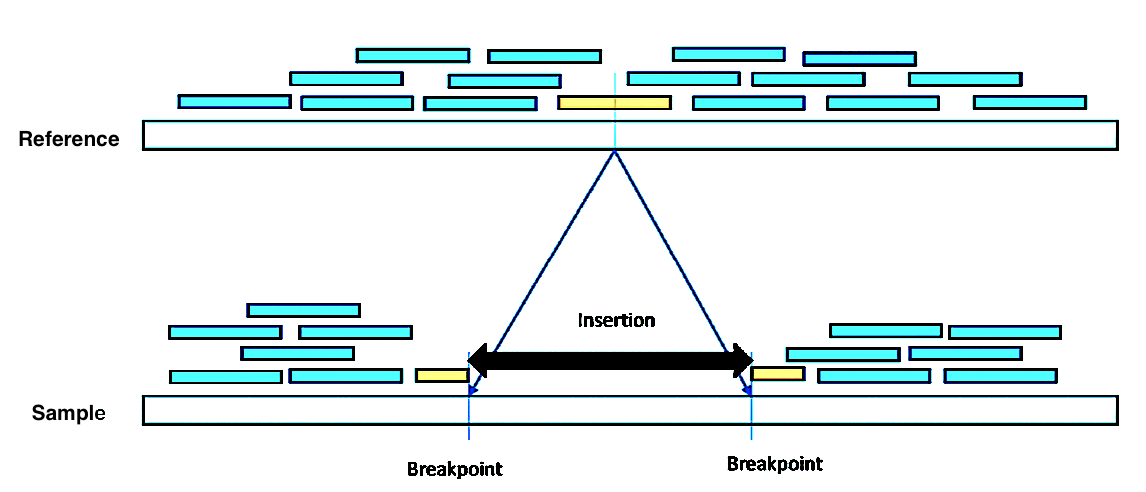
Figure 1: represents a deletion in the reference genome detected by split-read.
3. Hybrid/Multi-method structural variant calling approach
The De novo assembly and Paired-end approach specialize in determining specific types of SVs and not to identify all types and sizes of SVs. They rely on different properties of the underlying sequence data and vary in sensitivity and accuracy. Metamethods or a combination of methods can be used to serve the purpose of detecting all SV types. Higher efficacy yield in variant calling has been reported by using metamethods, such as:
Additionally, in the hybrid approach, usually, the SV calling process utilizes multiple variant callers to overcome the limitations of individual approaches. Knowing the advantages and drawbacks of various tools is imperative to make proper decisions when designing NGS SV calling data analysis pipelines. Some of the robust pipelines that integrate different structural variant callers are:
Concluding Remarks
In conclusion, SVs are gaining attention in evolutionary, population, and clinical genomics. This class of variants is increasingly being recognized as a significant source of genotypic and phenotypic variation. However, before SV calling becomes a routine in clinical diagnosis, challenges such as efficient detection of variants and correct genotyping using the results obtained from computational methods/algorithms must be overcome. In addition to this, another factor that complicates SV detection is the very nature of these variants, where their size and type differences make it much more difficult to assess their frequency using reference databases such as gnomeAD/ExAC. Whereas, the frequency assessment for SNVs is easier. Because of the former reason, it becomes arduous to determine if an SV variant occurs at a low frequency (< 0.5%) in the population and could be considered as a candidate for pathogenicity. Furthermore, the aforementioned lack of format standardization and metadata information has proved to be an issue in clinical applications since the need for quality assurance and diagnostic certifications is a must.
Overall, SV calling is on the verge of gaining importance with the rapidly evolving calling methods, however, the lack of benchmarks and reference databases ask for careful interpretation of these variants. With the fiercely competitive market of sequencing methodologies and their providers and the increase in the need for SV characterization, it will not take long for structural variant analysis to become a regular standard regime in clinical labs.
To learn more about gene prediction and how NGS can assist you, and to get access to all of our advanced materials including 20 training videos, presentations, workbooks, and private group membership, get on the Expert Sequencing wait list.

ABOUT DEEPAK KUMAR, PHD
GENOMICS SOFTWARE APPLICATION ENGINEER
Deepak Kumar is a Genomics Software Application Engineer (Bioinformatics) at Agilent Technologies. He is the founder of the Expert Sequencing Program (ExSeq) at Cheeky Scientist. The ExSeq program provides a holistic understanding of the Next Generation Sequencing (NGS) field - its intricate concepts, and insights on sequenced data computational analyses. He holds diverse professional experience in Bioinformatics and computational biology and is always keen on formulating computational solutions to biological problems.
More Written by Deepak Kumar, PhD









