How To Profile DNA And RNA Expression Using Next Generation Sequencing

Why is Next Generation Sequencing so powerful to explore and answer both clinical and research questions. With the ability to sequence whole genomes, identifying novel changes between individuals, to exploring what RNA sequences are being expressed, or to examine DNA modifications and protein-DNA interactions occurring that can help researchers better understand the complex regulation of transcription. This, in turn, allows them to characterize changes during different disease states, which can suggest a way to treat said disease.
Over the next two blogs, I will highlight these different methods along with illustrating how these can help clinical diagnostics as well as advancing our understanding of how to characterize and treat diseases. To start, we focus on methods to characterize the DNA and RNA directly. The next blog will look at how Next Generation Sequencing can be used in understanding modifications to the DNA, as well as characterizing protein binding regions of the DNA.
Let’s begin by learning more about sequencing DNA and RNA.
A. Genome and DNA sequencing methods
With the continued drop in sequencing costs, genome sequencing has become a powerful tool to expeditiously sequence and resequence genomes. The needs of the experiment will drive the level of sequencing that needs to be performed, which in turn impacts not just the cost (financially) of the experiment, but the time and effort (computationally) in analyzing the data.
1. Whole Genome Resequencing (WGR):
“Resequencing” is done to investigate the differences between the genome of specific individuals (ideally chosen based on phenotypes) and that of the reference genome. In other words, resequencing refers to the sequencing of the genome using Next Generation Sequencing methods for which reference genome is already available.
Consequently, the resequenced genome can then be compared with the reference genome to determine a catalog of mutations/aberrations (Single Nucleotide Variants, Copy Number Variants, Insertions, and Deletions) specific to each sequenced individual.
This method provides valuable insight into the individuals’ genetic background and hence helps in accurate clinical diagnoses.
2. Whole Genome De Novo Resequencing (WGDR):
When the reference genome is not available for downstream mutation determination analyses, the sequenced data obtained from resequencing is used to assemble a reference genome. These assembled genomes are used for downstream analyses and genes’ annotations.
This is mostly done for prokaryotes and viruses as their reference genomes are not readily available for research. WGDR is useful in metagenomics – the study of bacterial composition in environmental samples like wastewater/drinking water – done to identify the pathogenic bacterial strains. Such research is quite prevalent across labs in countries to improve water quality in the communities. The de novo genome assembly quality directly depends on library quality, sequencing accuracy, and sequencing coverage. The higher the coverage, the better the quality of the sequenced data. To better understand the essential concepts required for an efficient genome assembly, gene prediction and annotations, make sure to check out my other blogs.
3. Whole Exome Sequencing (WES):
Exons are the protein-coding regions that comprise less than 2% of the total genome but are enriched for disease-causing variants. Therefore, WES is more cost-effective to sequence than WGS, with many folds decrease in the effort and expense required for sequencing. WES has made it possible to conduct resequencing studies in species that have very large, repetitive, or polyploid genomes, such as some plants that have been selectively bred. There are a few significant exome-capture kits/methods available in the market – namely, HaloPlex, Ampliseq, SureSelect, and SeqCap. SureSelect and SeCap are “capture hybridization-based” as they rely on sonication of DNA fragments, followed by the hybridization of oligonucleotides specific to exons, whereas, HaloPlex and AmpliSeq are amplicon-based methods because they are based on PCR amplification of exonic regions using PCR primers (Figure 1).
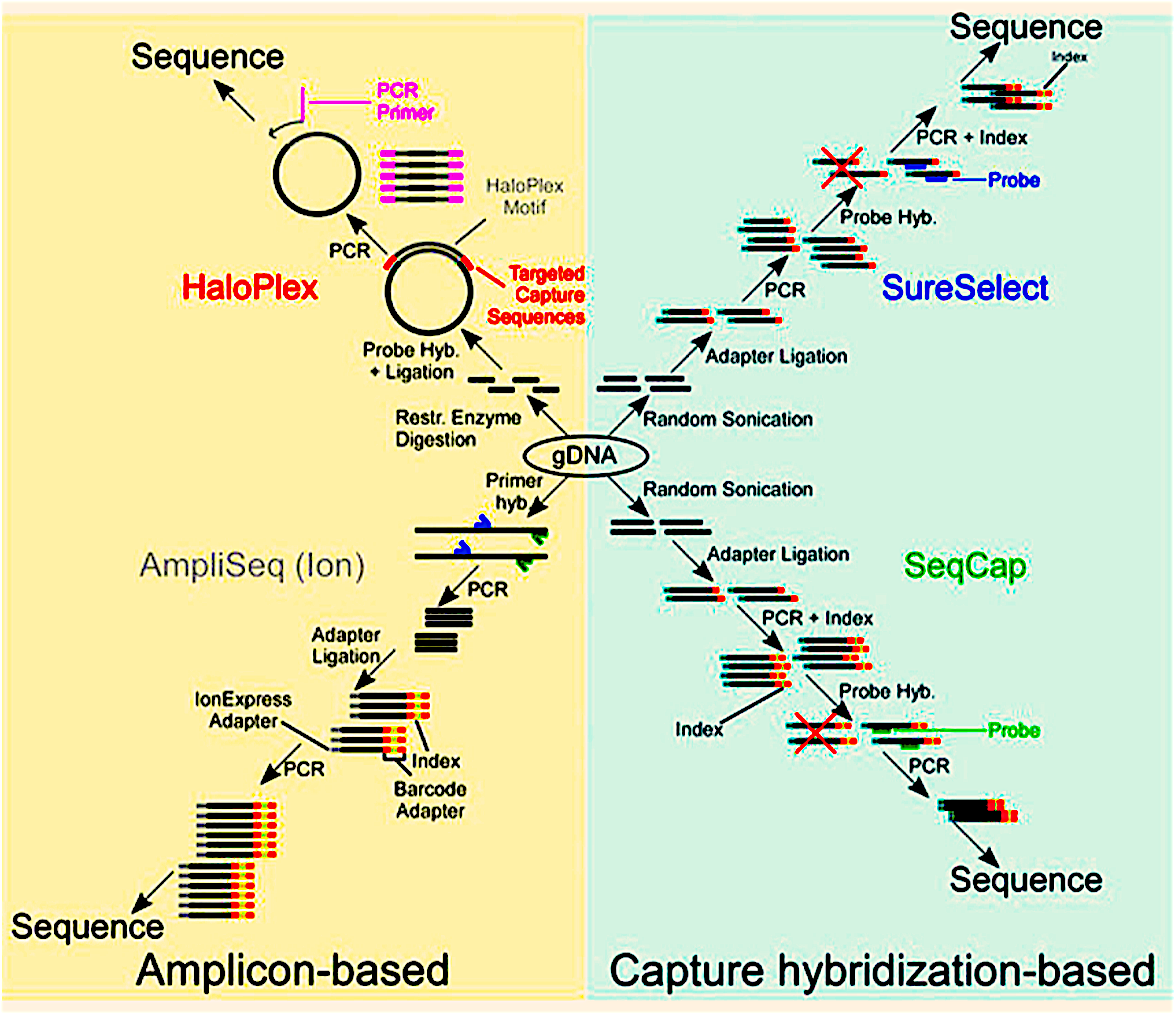
Figure 1: A representation of amplicon-based and capture hybridization-based exome capture kits.
4. Targeted Sequencing
Targeted sequencing determines the DNA sequence in a subset of genes or regions of the genome. Both WES and targeted sequencing focus time, expense, and data analysis on genomic regions of interest. WES is often confused with targeted sequencing as they are similar. The subtle difference is that WES characterizes all exons, but targeted sequencing characterizes only genomic regions of interest. An added advantage of targeted sequencing is it becomes more affordable to sequence at very high coverage, which is necessary for specific applications. For example, WGS typically provides 30-50x coverage, whereas targeted sequencing can cover the target region at 500-1000x. The higher coverage makes it possible to identify rare variants that would otherwise be undetected at lower coverage. Moreover, targeted sequencing generates a smaller and more manageable dataset, thus saving on the computational resources needed for analysis.
B. RNA Sequencing Methods
RNA sequencing or transcriptomic profiling can be used to perform high-throughput measurement of RNA levels and gene expressions. Compared to the microarray and Sanger sequencing-based approach, RNA sequencing is not limited in resolution, specificity, and sensitivity. Furthermore, prior knowledge of transcript sequences and their isoforms is not required.
Some advantages of RNA sequencing over established methods are:
—> The possibility of having a genome-wide coverage, regardless of whether a reference genome is available.
—> Better sensitivity and specificity. The dynamic range of RNA-Seq spans five orders of magnitude given sufficient coverage, which is significantly higher than the existing array-based measurements, which can measure at most three orders of magnitude in changes.
—> Unbiased detection of all transcripts and isoforms, including novel ones.
—> Detection of transcripts or isoforms expressed at low levels if the sequencing is done at sufficient depth.
—> Absence of background noise from microarray hybridization.
—> Detection of genetic variants that affect transcripts, especially for highly and moderately expressed ones.
In cancer research, gene expression and small RNA profiling provide information about active pathways that drive tumorigenesis, which may not be captured with genome sequencing. The Cancer Genome Atlas Project has profiled the transcriptomes of over 4,000 cancer tissue samples in 12 cancer types. Also, clinical tests that use RNA-Seq, such as OncotypeDX, which predict drug responses and individual prognosis based on gene expression signature, are now commercially available in the US.
Beyond simply profiling RNA molecules, RNA-Seq is a great tool to help us better understand epigenetics and RNA biology, especially with the advent of innovative techniques. For example, RNA-Seq methods could be used to capture snapshots of alternative splicing, RNA editing, nascent transcripts, ribosome-bound transcripts, and fusion transcripts.
Apart from RNA and mRNA profiling, combinations of molecular biology and biochemical techniques with Next Generation Sequencing have led to the development of many RNA-Seq derived methods:
a. Ribo-Seq Ribosome profile sequencing: to identify RNAs being processed by the ribosome to monitor the translation process
b. miRNA-Seq: Sequencing for microRNAs
c. ChIRP-Seq (Chromatin Isolation by RNA): purification – to discover regions of the genome bound by specific RNA
d. PAR-CLIP (Photoactivatable-Ribonucleoside): Enhanced Crosslinking and Immunoprecipitation sequencing – to identify and characterize binding sites of RNA-binding proteins and miRNA-containing ribonucleoprotein complexes (miRNPs)
e. CLIP-Seq (Cross-linking and Immunoprecipitation Sequencing): to identify the binding sites of cellular RNA-binding proteins (RBPs) using UV light to cross-link RNAs to RBPs
Concluding Remarks
Taken together, the sequencing of DNA at the whole genome level to just the whole exon-level can provide the researcher with a tremendous amount of data that can help identify markers for a disease, which can be further expanded to look at what RNA transcripts are present as a result of these marker sequences. Next Generation Sequencing really offers the ability to decode the book of life and figure out where misprints lead to adverse outcomes.
In future blog articles, we will delve into the finer details of these methods and why you would choose one method over another. Tune back in next time when we will explore how Next Generation Sequencing can be used in understanding DNA modification and identifying sites of DNA:Protein interaction
To learn more about gene prediction and how NGS can assist you, and to get access to all of our advanced materials including 20 training videos, presentations, workbooks, and private group membership, get on the Expert Sequencing wait list.

ABOUT DEEPAK KUMAR, PHD
GENOMICS SOFTWARE APPLICATION ENGINEER
Deepak Kumar is a Genomics Software Application Engineer (Bioinformatics) at Agilent Technologies. He is the founder of the Expert Sequencing Program (ExSeq) at Cheeky Scientist. The ExSeq program provides a holistic understanding of the Next Generation Sequencing (NGS) field - its intricate concepts, and insights on sequenced data computational analyses. He holds diverse professional experience in Bioinformatics and computational biology and is always keen on formulating computational solutions to biological problems.
More Written by Deepak Kumar, PhD











