How To Determine The Optimal Antibody Concentration For Your Flow Cytometry Experiment (Part 1 of 6)

Over the next series of blog posts, we will explore the different aspects of optimizing a polychromatic flow cytometry panel. These steps range from figuring out the best voltage to use, which controls are critical for data interpretation, what quality control tools can be integrated into the assay; how to block cells, and more. This blog will focus on determining the optimal antibody concentration.
As a reminder about the antibody structure, a schematic of an antibody is shown below.
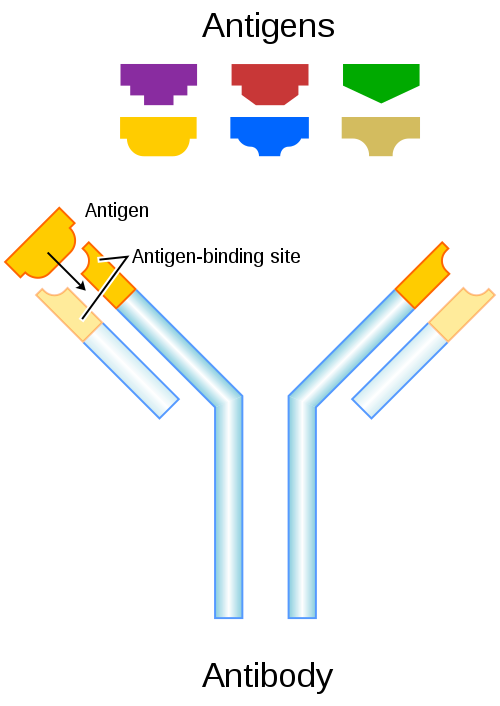
The antibody is composed of two heavy chains and two light chains that are joined together by disulfide bonds. At the top is the antigen-binding site, and at the bottom is the constant region of the antibody. When digested with a proteolytic enzyme such as papain, the antibody will separate into two Fab fragments, and the tail, or Fc fragment.
There are three different types of binding interactions that an antibody can undergo.
The first is the antibody binding to its target antigen. This is a specific and reversible binding that obeys simple kinetics:
ka
[R] + [L] ⇄ [RL]
kd
The ka is the rate of formation, while the kd is the rate of dissociation. There are various tools that can be used to measure this interaction.
The second binding interaction is a specific reaction between the Fc fragment and one of several types of Fc Receptors that are expressed on certain cells. This is a specific interaction, but generally not something that we are interested in. Minimizing this interaction will be the topic of a future blog post.
The third binding interaction is what is termed non-specific binding. This is when the Fab fragment binds to a low-affinity target. This binding is responsible for the increase in background staining of a flow cytometry experiment.
In flow cytometry, the goal is to make the most sensitive measurement we can make. There are many things that reduce the sensitivity of our measurements including autofluorescence, spectral spillover, and increasing background. This is where titration comes into play. The goal of titration is to identify the best optimal antibody concentration that results in the highest signal to noise measurement. In this process, we will label cells over a concentration range, calculate the staining index (SI) at each point, and plot concentration vs SI.
The first question that is often asked is how many points should you use? An informal survey was conducted and the results are:

There is no clear consensus in the flow cytometry community as to how many points should be done. This survey suggests ‘5 points’ is used by a large percentage of researchers.. The crucial point of the titration experiment is to ensure that you identify the optimal signal to noise and have some points around that value.
When you perform the titration experiment, make sure to follow the protocol that you would be using the antibody in. For example, if you are performing intracellular staining, make your titration reflect that. Likewise, you should consider adding a viability dye to remove any dead cells that can confound the results by increasing the cv of the data. There are two schools of thought in this matter, one suggests to use the same viability dye as the one you will use in the experiment, the other suggestsy to use a viability dye that will not impact the staining of the target (i.e. require compensation).
A typical titration curve is shown below in figure 3.


For antibody A, the maximal separation is somewhere between 0.25 and 1 micro liters. In this case, the researchers chose 0.5 micro liters. Why? At this concentration, if there are any issues in the preparation (i.e. pipetting errors), a little less or a little more will not change the SI. However, at 0.25 micro liters if there is an issue, and less antibody is used, the SI decreases, which could impact the resolution of the data. Regarding antibody B, the concentration of 0.25 micro liters was chosen for the same reason.
Recalling the equation above about the kinetics of the reaction, we can take advantage of biochemistry to calculate the binding kinetics, assuming Michaelis-Menten kinetics of binding. In this case, we would plot the antibody volume vs median fluorescent intensity and calculate the best fit line to this data. From that, we can extrapolate the Vmax and KM values. Assuming that the saturation is at 80%, it is possible to extrapolate the antibody concentration. This data is shown below, calculated in FCS Express, with their built-in regression toolkit.

As you will recall, for antibody A, the SI suggested 0.5 micro liters, and for antibody B, 0.25 micro liters.
Which to choose? Which is correct? Interestingly, the answer is ‘it depends.’ The purpose of the staining index is to look at the best separation between the background and positive signal, which is what is most important in the experiment. In fact, in the Cyto2015 tutorial ‘Forensic Flow Cytometry’ on slide 31, the authors state that ‘there is often at least a 4x range of concentrations that are just fine’ and they show it on the next slide to make their point. If you are interested in reading more about this topic, we recommend a recent publication by Kalina, Lundsten and Engel.
At the end of the date, it is important to use optimal antibody concentration, with an eye to ensuring that there is the best separation between positives and negatives, which will help ensure the most sensitive measurement is being made in the experiment.
To learn more about important control measures for your flow cytometry lab, and to get access to all of our advanced materials including 20 training videos, presentations, workbooks, and private group membership, get on the Flow Cytometry Mastery Class wait list.

ABOUT TIM BUSHNELL, PHD
Tim Bushnell holds a PhD in Biology from the Rensselaer Polytechnic Institute. He is a co-founder of—and didactic mind behind—ExCyte, the world’s leading flow cytometry training company, which organization boasts a veritable library of in-the-lab resources on sequencing, microscopy, and related topics in the life sciences.
More Written by Tim Bushnell, PhD











