Best Flow Cytometry Cell Sorting Practices

Walk into any flow cytometry facility and you will see one or more cell sorters. These devices use the principles of flow cytometry to isolate phenotypically defined cells to a high degree of purity for any of a number of downstream applications. Even single cells can be isolated for cloning and single-cell genomics analysis, a very hot area of research these days.
This was not always the case. Prior to 1965, if a researcher wanted to isolate cells, their only choice was some form of gradient centrifugation, a bulk separation method. There were no real options for anything with more fine control.
That changed when Mack Fulwyler published this paper in which he described an instrument capable of measuring an object’s Coulter volume and isolating the cells based on this volume. The ingenious part of the system was the use of the technology that Richard Sweet had developed for the “ink jet oscillograph”. This first cell sorter is shown in Figure 1.

Figure 1: One of the first cell sorters built by Mack Fulwyler.
Fulwyler demonstrated, using a mixture of mouse and human erythrocytes, that these 2 populations could be separated, as shown in Figure 2 from that paper.
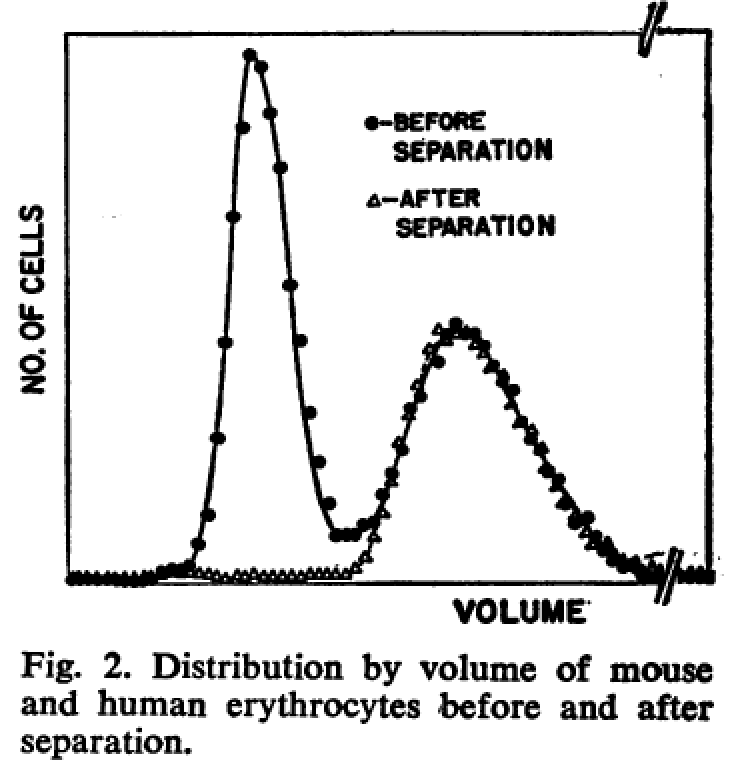
Figure 2: Results from Fulwyler’s paper, showing the separation of a mixture of cells.
This was revolutionary and led to the technology available today, where researchers can isolate very specific populations of cells, to have homogeneity from a heterogeneous population, and then do downstream applications with those cells.
Enter Len Herzenberg, who coined the term, FACS — or “fluorescent activated cell sorting” — who was one of the first biologists to see the power in this new invention. He took Fulwyler’s plans and, with the assistance of the engineers at Stanford’s Instrumentation Research Laboratory, added an arc lamp (1969) and later an argon laser (1972). The race was on as Becton Dickinson took these ideas and began marketing the first cell sorters.
Cell sorting is a central tool to many experiments, so it is only right to look at some strategies that can be used to get high-quality results from a sorting experiment. The focus of these strategies is on electrostatic cell sorters.
1. Find your ideal sample concentration.
The correct input sample concentration is critical to ensure that your cells are delivered in a consistent concentration to the sorter. Too many cells per second, and the purity and yield are compromised. Too few cells per second, and the sort can take a long time, potentially impacting the quality of the sorted cells.
The proper concentration is a combination of the cell type you are using and the nozzle size of the cytometer. The goal is to have a nozzle that is about 5 times the size of the cells being sorted. The larger the cell, the larger the nozzle, which in turn impacts the pressure of the sheath fluid as well as the frequency of droplet generation. Below, is a rough guide based on some common cell volumes based on data from this site.

Figure 3: Approximate cell diameters for different cell types and recommended nozzle size.
The next step is to understand the the cell sorter actually sorts droplets, not cells. These droplets are generated by a pizoelectric element that vibrates the stream at some frequency, and the smaller the nozzle, the higher the frequency. Based on Poisson statistics, we would like to have an event rate of approximately 1 cell per 4 droplets. This event rate gives us the greatest probability that there is not a cell in the leading or lagging droplet, while maintaining a reasonable sorting rate. Shown in Figure 4 are some recommended values for sorting rate and cell concentration. Of course, check with the sort team to ensure you know the values for your specific instrument.

Figure 4: Recommendations for event rate and cell concentration, based on nozzle size and droplet generation frequency.
If the sample is too concentrated, you’re going to increase the abort rates, which occur when a second cell enters the analysis window before the first cell is finished processing. In this case, the data for both cells is lost. Further, if you have too many events per second, the Poisson statistics become skewed, increasing the probability of both 2 cells in one droplet, or cells in the leading and lagging droplet. The result of this compromises your ability to sort and get a good purity and recovery.
Of course, this sort of data can also be used to estimate the time it will take to sort the sample. This is shown in Figure 5.

Figure 5: Time to sort 100,000 cells, based on population frequency and droplet frequency.
This data is from one facility, so ask your local facility if they can provide this information for you. It will help you plan your experiment in more detail and help determine how long a sort will take.
2. Use magnetic sorting to enrich a rare population.
What if, based on the calculations above, your sort is going to take too long? This usually happens with rare event sorts. For example, as Figure 5 shows, if 100,000 cells of a 0.1% frequency are needed, the sort could take from 2.5 to 11 hours, depending on cell size. If more cells are needed, it will take even longer. How can one reduce the sort time?
Enter magnetic cell sorting. With this technique, you label your cells with antibodies coupled to a magnetic bead, and expose the solution to a magnetic field. Cells with the magnetic bead are retained while the other cells remain in the supernatant. While magnetic separation was used for years, in this paper the MACS method was described. The MACS system uses a paramagnetic bead and a column for the separation. Other methods do not employ the same bead and can be done in solution. The choice is up to the investigator.
Magnetic separation is a great pre-enrichment step for cell sorting, and sometimes it may even be sufficient for what you need. For example, if you need CD4 cells, magnetic sorting may be all you need to do with your blood to isolate your CD4 cells.
But in general, it’s used for pre-enriching the sample.
Here’s an example: there are 100 million bone marrow cells, the population of interest is 0.1%, the sort speed is at 15 million cells an hour, so it’s going to take you 2 hours to sort that population.
However, if you perform some sort of pre-enrichment, such as using a magnetic storage particle bead that enriches the population, it’s possible to go from that larger population of 100 million cells down to an enriched population of about 10 million cells. Enriching the population also means that you will be sorting the population at a higher frequency than 0.1%.
Sorting 10 million cells on a machine that can sort 50 million cells an hour means it takes about 20 minutes, instead of 2 hours. The time saved can get even more significant depending on the frequency and how many cells you are starting with. The end result is that the cells are happier, as is the researcher.
3. Suspend cells in the right buffer to avoid cell clumps.
Flow cytometry is a single-cell technique, so having a good single-cell suspension is critical for good results when sorting. Additionally, since the nozzle is smaller than it is on a typical analyzer, clumps will lead to clogs, which slow down the sort and cause unnecessary grief.
It is critical to examine your cells under a microscope before sorting. See how good the single cell suspension is. Also, you should filter just before sorting to remove any clumps in the solution. Beyond that, remove aggregates from the sort gate with doublet discriminator. You can learn more about doublet discrimination in this blog.
The buffer can make all the difference in the quality of the single-cell suspension. If, for example, you are working with non-adherent cells, a buffer of PBS with 0.5% BSA is a good basic buffer. Adding 25 mM HEPES buffer (pH 7.0) is a good idea as well, as HEPES has better buffering properties at high pressure than PBS does. Finally, a smidge (1 mM) of EDTA is good to add, as it helps chelate divalent cations that are often required for the formation of cell aggregates.
If you are working with lymphocytes, you can often omit the EDTA. If you have a high percentage of dead cells, adding 10 units/ml of DNAse II is strongly recommended. This will help reduce clumping caused by free DNA.
Finally, adherent cells require additional efforts. This can include increasing the EDTA to 5 mM, but be careful as too much EDTA can be bad for cells. Likewise, if you are adding DNAse, it requires Mg++ to work and EDTA will reduce/inactivate the DNAse. Consider neutralizing trypsin used to remove the cells with BSA or trypsin inhibitors, rather than FBS.
On the other end of the system, the catch buffer needs to be considered. Your collection buffer needs to be designed for your cells to enhance viability.
It is not advisable to use 100% FBS as a collection buffer, for 2 reasons. First, it is a higher density than the cells so, when the cells start to hit it, they are not actually mixing in, so they’re not actually loading up the FBS. Second, the few cells that get in the FBS early on are exposed to 100% FBS, so they are getting huge doses of whatever else is in the FBS and this will affect the cells.
The most recommended collection buffer is your cell culture medium with 10% FBS or some other serum.
Don’t forget antibiotics, pen-strep or gentamycin, and antifungal agents — you don’t want to introduce any contamination.
4. Change your instrument settings when sorting small cells.
Small cells, bacteria cells, and things of similar size, test the limits of resolution of most cell sorting systems. Most systems have been designed for looking at mammalian cells, so if you’re looking at a 0.3-micron bacteria, it’s much harder to find in the background noise.
Beads can help to calibrate your instrument for the small particle detection, as can a forward-scattered PMT. Side scatter may be a better option here, especially if it is possible to use a lower wavelength (405 nm, for example), as this may also improve resolution.
Don’t forget to change your data collection to log scale. Better still, it’s best not to rely upon scatter at all. Fluorescence is a much better trigger and there are a variety of fluorescent dyes that will stain live bacteria which can help identify your bacterial population of interest.
You can also back gate on the fluorescence to see where the populations are in your other plots. You aren’t going to get the resolution discrimination that you would have on, say, a population of lymphocytes from PBMCs.
Finally, with small cells, core stream size is important, as is the event rate. Keep to the recommended sorting rates as described above.
5. Optimize your sample preparation and instrument when sorting large cells.
What about sorting big cells? Slow it down.
Based on the data above, you are going to have to use less concentrated samples with the larger nozzle.
Interestingly, when using the 130 micron nozzle, ear protection may be required as the drop drive frequency can enter into the range of human hearing, and it’s annoying at the minimum.
DNAse is also a must, especially with a larger sample, because they are more fragile and easier to break. Easy to break means more DNA coming out, and more DNA means more things getting stuck together.
The collection buffers for large cells also have to be heavily optimized to preserve the larger cells.
Now, what about really, really, really large cells? Such as this C. elegans that has been transfected with GFP?
Figure 6: C. elegans transfected with GFP tag.
You can see spots of GFP. If you wanted to sort this C. elegans from a C. elegans that didn’t have spots, what could you do?
This requires a special instrument, which is shown below. Made by Union Biometrica, the BioSorter Platform is perfect for very large cells and small organisms.

This system has a range from 10 to 1,500 microns, so this system will sort neurospheres, adipocytes, lipid bodies, and other things that are large fragile cells. It will sort C. elegans, and it will sort fly larvae.
Cell sorting by flow cytometry is a powerful method first developed by Mack Fulwyler. Today’s researchers have access to machines that can sort up to 6 populations simultaneously with many fluorochromes used to finely subset the cell populations. It is a central technology that serves as a gateway to additional techniques, from cell culture to generating xenograph models, to genomic analysis.
As a researcher, you want to achieve the best cell sorting possible. So, how can you achieve that? There are clear strategies you can use to achieve great cell sorting results, including finding your ideal sample concentration, using magnetic sorting to enrich your population, suspending cells in the right buffer to avoid cell clumps, changing your instrument settings when sorting small cells, and optimizing your sample preparation and instrument when sorting large cells. Happy sorting.
To learn more about Best Flow Cytometry Cell Sorting Practices, and to get access to all of our advanced materials including 20 training videos, presentations, workbooks, and private group membership, get on the Flow Cytometry Mastery Class wait list.

ABOUT TIM BUSHNELL, PHD
Tim Bushnell holds a PhD in Biology from the Rensselaer Polytechnic Institute. He is a co-founder of—and didactic mind behind—ExCyte, the world’s leading flow cytometry training company, which organization boasts a veritable library of in-the-lab resources on sequencing, microscopy, and related topics in the life sciences.
More Written by Tim Bushnell, PhD











